
Transcriptome analysis is a crucial step in understanding gene expression and regulation in biological and disease studies. However, traditional RNA sequencing methods have limitations in resolving complex transcript structures, such as those generated by alternative splicing. This is partly because these technologies use short read sequencing, which is less successful in resolving difficult sequencing regions.
In order to support short read sequencing methods, standard library preparation protocols include fragmentation steps which shear full length cDNA, yielding smaller molecules which are more suitable for short read sequencing. These short fragments, however, require transcript assembly to combine reads into a full length molecule.
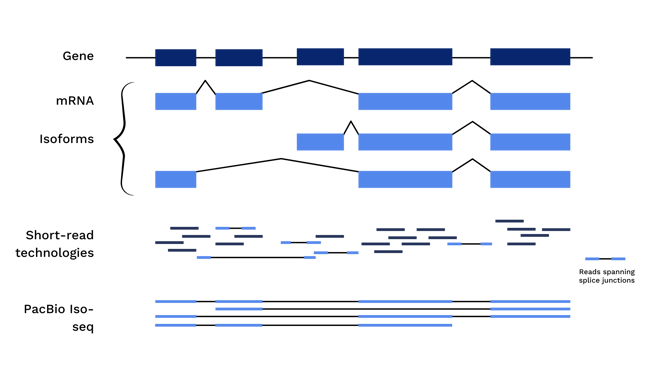 As a result, RNA processing steps, like alternative splicing or fusion events, cannot be accurately detected using short read sequencing. A short piece of an exonic region could be present in one or many isoforms, or in a gene with or without a fusion event. Long read sequencing, however, removes the need for fragmentation and facilitates sequencing of the entire length of the cDNA molecule, including all exonic regions and fusion sites.
As a result, RNA processing steps, like alternative splicing or fusion events, cannot be accurately detected using short read sequencing. A short piece of an exonic region could be present in one or many isoforms, or in a gene with or without a fusion event. Long read sequencing, however, removes the need for fragmentation and facilitates sequencing of the entire length of the cDNA molecule, including all exonic regions and fusion sites.
Why Does Long Read RNA Sequencing Matter?
The complexity of an organism’s transcriptome and proteome is only in part due to its genome. While the DNA sequence provides the building blocks for transcription and translation, how those building blocks function is due to a variety of regulation steps which occur in a cell. This can be a direct action on the DNA itself, such as methylation to activate or inactivate transcription. It can also be a modification of the transcribed RNA, as occurs with alternative splicing.
Looking at the DNA sequence of a gene does not determine the final sequence of the corresponding RNA molecule, nor does it identify the specific structure and function of the translated protein. Alternative splicing and its resulting isoforms enable a single gene to code for a variety of proteins, yielding a much more diverse proteome than suggested by a limited number of protein coding regions in the DNA.
To fully understand a cell’s behavior, it is not enough to know which regions of DNA are transcribed. Research must delve into the variations in those RNAs, triggered by splicing events. Short read sequencing may show high levels of expression of a single exon. But in actuality, high expression may correspond to a multitude of gene isoforms due to alternative splicing, different transcription start sites, or untranslated regions (UTRs).
When re-combining short transcript sequencing reads, reads can be aligned to known regions, but they cannot always be accurately spliced together to re-create a single molecule. Thus the true picture of a complete transcript may not be accurate.
Full length cDNA sequencing, however, yields a picture of an mRNA transcript from its 3’ polyA tail through the 5’ transcription start site, identifying splice junctions, retained introns, and UTRs along the way. Diseases, including cancer, can be the direct result of changes to alternative splicing mechanisms, yielding gain or loss of function mutations and changes in downstream protein expression. Identifying isoform changes can lead to targeted approaches for disease treatment and understanding.
Two RNA Technologies: MAS-Seq and Iso-Seq
The first methods for high throughput, third generation, full length cDNA sequencing arose almost a decade ago with the advent of PacBio’s Iso-Seq methodology. Iso-Seq methods require synthesis of a full length cDNA molecule, taking advantage of template switching oligos (TSO) which function as a primer for second strand cDNA synthesis and amplification. cDNA molecules are used for Iso-Seq rather than a direct RNA approach, as the cDNA molecule is far more stable during subsequent library prep and sequencing steps.
Iso-seq library preparation incorporates the PacBio specific SMRTbell adapters, yielding a sequencing ready molecule. Each library molecule contains one transcript which can be captured in a PacBio SMRT cell zero mode waveguide (ZMW). Because cDNA molecules have an average size of only 1.5-2kb (ranging from ~400-9000bp), Iso-seq technologies do not take full advantage of the long read capabilities of the PacBio SMRT cells.
Recent advances in the Iso-seq methods paved the way for the new Kinnex protocols from PacBio. Kinnex quite literally combines the full length cDNA generation of TSO enabled protocols with the long read capabilities of the SMRTseq technology. Kinnex library preparation methods use a simple PCR reaction to add small adapters to the end of full length cDNA molecules. The adapter on one transcript anneals to the adapter on another, yielding a chain of cDNA molecules connected by short, known sequences.
The final SMRTbell library molecule contains up to 12 different full length transcripts, allowing each sequencing ZMW to yield 10-12 different transcript sequences, essentially increasing data output 12-fold from the same SMRTcell compared to Iso-Seq methods. Kinnex is adaptable for bulk RNA, 16s, and 10x generated single cell cDNA.
In the Field
MAS-Seq and Iso-Seq capabilities are already making progress in rare Mendelian disease research. In 2023, researchers teamed up with PacBio scientists to develop a multiomic approach to understanding the molecular basis of undiagnosed diseases.
The group applied this analysis of the genome, methylome, epigenome, and transcriptome to a participant in the Undiagnosed Diseases Network. With MAS-Seq, they were able to identify the disruption of four genes, each by a distinct mechanism. Without the combination of multiple omics technologies and the benefit of long reads, it had been nearly impossible to identify all of these mutations.
Iso-Seq is also particularly useful in plant research, where genomes tend to be much larger than the human genome. A 2017 study out of Malaysia used Iso-Seq to map the transcriptome of three unique pitcher plant species. Scientists compared the transcriptomes of carnivorous, non-carnivorous, and a hybrid cross of the two species, permitting the team to uncover insights into the unique traits of carnivorous plants. At the time, there was limited genetic information about this plant family. This study generated full reference transcriptomes for all three plant specimens.
PacBio Iso-Seq is particularly valuable in fields like genomics, transcriptomics, and functional genomics. It can provide insights into gene structure, alternative splicing, gene expression, and facilitate the discovery of novel transcripts. Long reads and full-length transcript information make it a powerful tool for characterizing complex transcriptomes and identifying rare or low-abundance transcripts.





